Jan 2020
Rhabdomyosarcoma
Reviewer(s): Jayalakshmi Venkateswaran, MD; Dharam Ramnani, MD
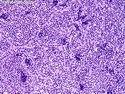
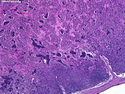
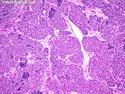
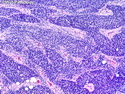
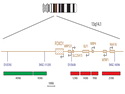
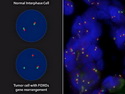
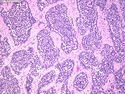
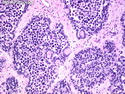
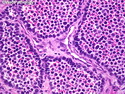
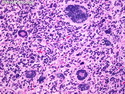
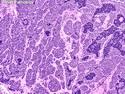
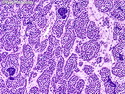
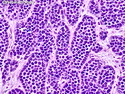
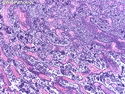
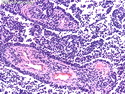
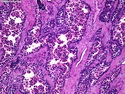
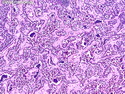
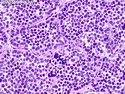
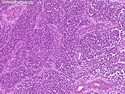
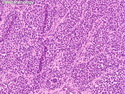
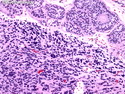
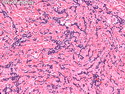
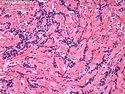
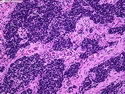
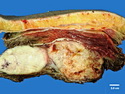
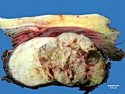
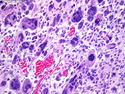
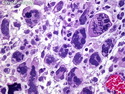
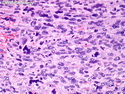
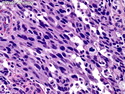
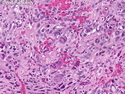
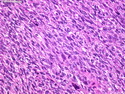
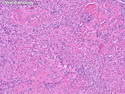
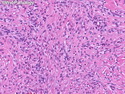
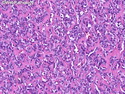
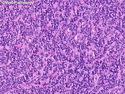
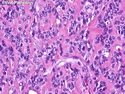
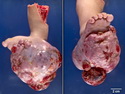
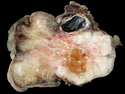
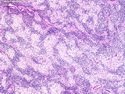
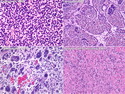
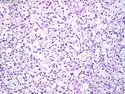
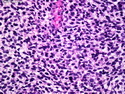
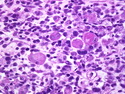
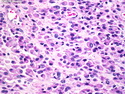
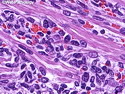
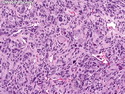
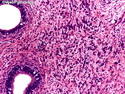
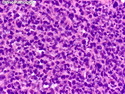
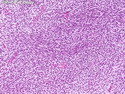
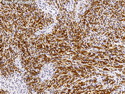
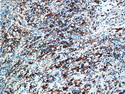
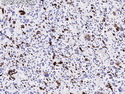
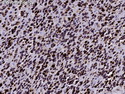
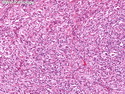
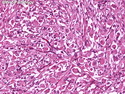
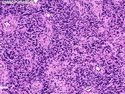
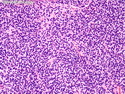
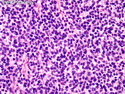
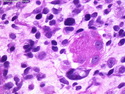
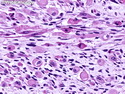
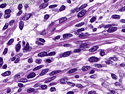
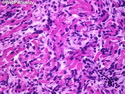
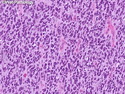
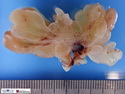
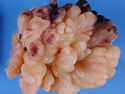
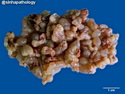
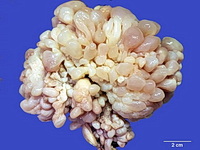
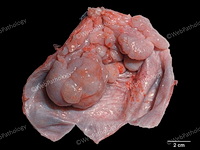
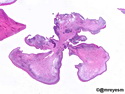
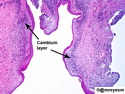
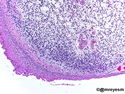
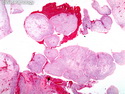
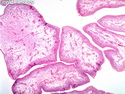
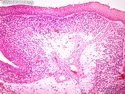
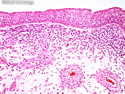
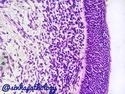
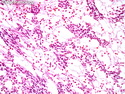
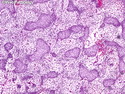
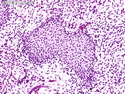
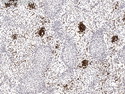
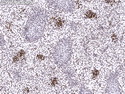
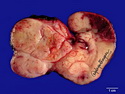
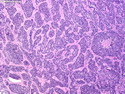
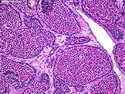
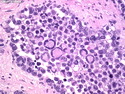
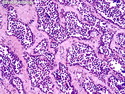
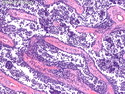
Rhabdomyosarcomas (RMS) are malignant soft tissue tumors, exhibiting skeletal muscle differentiation. There is a bimodal age distribution, between 2-6 years and a second peak between 10-18 years; it is uncommon after 45 years. Common locations include head and neck (26%), genitourinary tract (17%) and extremities (15%). There are four major subtypes - Embryonal, Alveolar, Pleomorphic, & Spindle Cell/Sclerosing. Sarcoma botryoides is a variant of Embryonal RMS. Grossly, their size is variable and they may be well-circumscribed and multinodular or less well defined with infiltrative borders. Sectioning reveals glistening, gelatinous (mucoid) or gray-white or pink-tan fleshy and bulging appearance. Areas of cyst formation, hemorrhage or necrosis may be present. Microscopically, the tumor cells range from small primitive hyperchromatic round/spindle/stellate cells to large differentiated strap/ribbon/tadpole/racket-shaped cells with abundant eosinophilic cytoplasm and cross-striations (rhabdomyoblasts). The nuclei are large, round, and vesicular with a prominent nucleolus. Immunohistochemically, they are positive for MyoD1, Myogenin A, Muscle Specific Actin (MSA) or Desmin. Translocation t(2;13)(q35;q14) is found in the majority of alveolar RMS cases and a t(1;13)(p36;q14) is noted in a smaller subset. The congenital/infantile form of sclerosing/spindle cell RMS has gene fusions involving VGLL2 and NCOA2 or CITED2 genes. Usually RMS is treated with surgery and chemotherapy, with or without radiation therapy. The histologic type and location of the tumor influence survival. The botryoid variant of embryonal RMS has the best prognosis, while the pleomorphic subtype is often fatal. References:1) Goldblum, J. R., Weiss, S. W., & Folpe, A. L. (2019). Enzinger & Weiss's Soft Tissue Tumors - Seventh Edition. Philadelphia, PA. Elsevier. 2) Goldblum, J. R. et al (2018). Rosai and Ackerman's Surgical Pathology - Eleventh Edition. Philadelphia, PA. Elsevier.
.jpg)
.jpg)