May 2020
Acinar Cell Tumors
Reviewer(s): Dharam Ramnani, MD
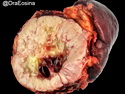
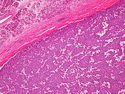
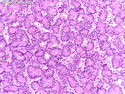
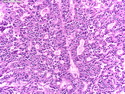
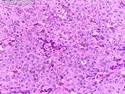
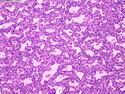
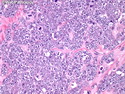
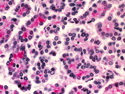
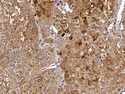
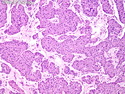
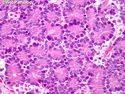
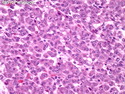
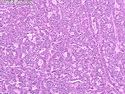
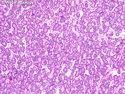
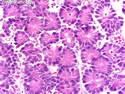
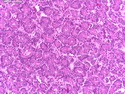
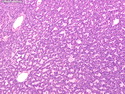
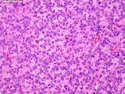
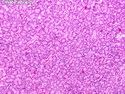
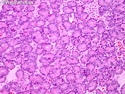
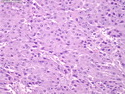
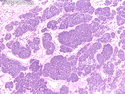
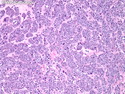
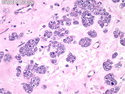
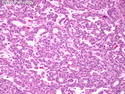
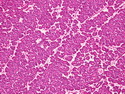
Acinar cell carcinoma (ACC) is a rare high-grade neoplasm of pancreas with acinar differentiation. It makes up 1%-2% of all pancreatic neoplasms in adults and 15% in children. Most cases are sporadic. Some are associated with Lynch syndrome, Carney complex, or familial adenomatous polyposis. Symptoms include abdominal pain, nausea, vomiting, and weight loss. Jaundice is rare. AFP levels may be elevated. About 15% of patients (usually with liver metastases) develop a lipase hypersecretion syndrome (peripheral fat necrosis, polyarthralgias, and eosinophilia). Mutations in KRAS, SMAD4, and TP53 are rare. Small % of cases show: CDKN2A or CDKN2B mutations; alterations in the APC/β-catenin pathway (mutations of APC and CTNNB1); rearrangements involving BRAF and RAF1; MSI (Lynch syndrome) ; alterations in MYC (gene amplification, chromosome 8 polysomy). 18q del with loss of DCC is an early event. ACC is well-circumscribed, partially encapsulated, and large solid tumor (8-10 cm average) with necrosis and cystic degeneration. Cells show acinar, solid, glandular, or trabecular patterns. Rare case have oncocytic, spindle cell, clear cell, or pleomorphic features. Cells have large vesicular nuclei, prominent nucleoli, and PAS-D positive zymogen granules. Mixed tumors with ACC and neuroendocrine (NE) or ductal components do occur. Useful markers include trypsin, chymotrypsin, lipase, and BCL10. NE markers are often focally positive. ACC is an aggressive malignancy. Prognosis is slightly better than pancreatic ductal adenocarcinoma. The 5-yr survival rate for resectable and unresectable tumors is 72% and 22% respectively. References:WHO Classification of Tumours Editorial Board. Digestive System Tumors, 5th Edition, 2019; IARC, Lyon, France. Goldblum, J. R. et al (2018). Rosai and Ackerman's Surgical Pathology, 11th Edition. Philadelphia, PA. Elsevier.