
Chordoma : Pathogenesis & Molecular Changes



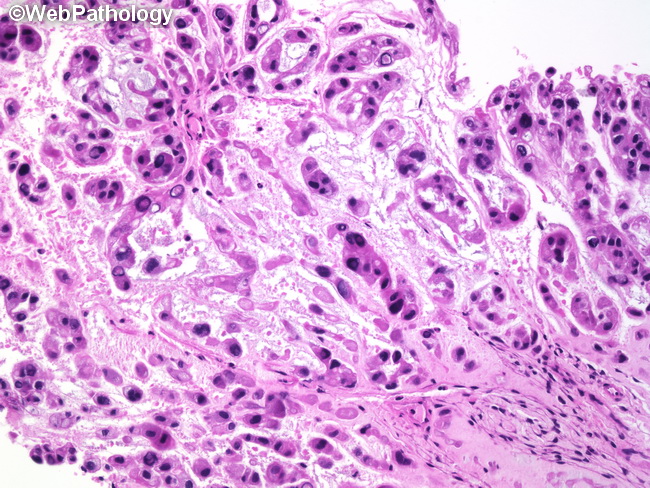
Comments:
Chordoma - Pathogenesis & Molecular Changes: Benign notochordal tumor, which arises from notochordal remnants, has been suggested by some as a possible precursor of chordomas; however, this area remains controversial. Brachyury - a transcription factor encoded by TBXT gene and belonging to the family of T-box proteins - is involved in notochord development and maintenance. Genetic alterations in TBXT gene play a role in the pathogenesis of chordoma. Familial chordomas show TBXT gene duplications with upregulation of brachyury, whereas about 5% of sporadic cases show TBXT gene amplification and single nucleotide polymorphisms. Rare childhood chordomas are associated with tuberous sclerosis caused by germline loss-of-function mutations in the tumor suppressor gene TSC1 or TSC2. Tyrosine kinase receptors, including EGFR, PDGFR-alpha, and PDGFR-beta have been implicated in the pathogenesis of chordomas. Chordomas show abnormalities in several molecular signaling pathways such as PI3K/AKT/mTOR (16% cases), MAP/ERK pathway, JAK/STAT pathway, and retinoblastoma pathway. This knowledge has been exploited in creating novel molecular therapies that target specific genetic lesions. In addition, several mutations have been reported in chordomas, including ALK, CTNNB1, NRAS, PTEN and CDKN2A. The de-differentiated variant shows TP53 mutations. Poorly-differentiation chordomas harbor loss of chromosome 22q and SMARCB1. About this image: Chordomas arising in the spheno-occipital region (clivus) (shown here) often cause symptoms referable to compression of cranial nerves (especially optic nerve) and pituitary gland. Some cases extend inferiorly causing nasal obstruction. The appearance is typical of chordomas arising elsewhere. The tumor cells have abundant eosinophilic cytoplasm with moderate nuclear atypia and form anastomosing cords. Chordomas arising in this location tend to present on an average about a decade earlier than those arising in the sacrococcygeal region.