
Fabry Disease : Genetics & Clinical



Comments:
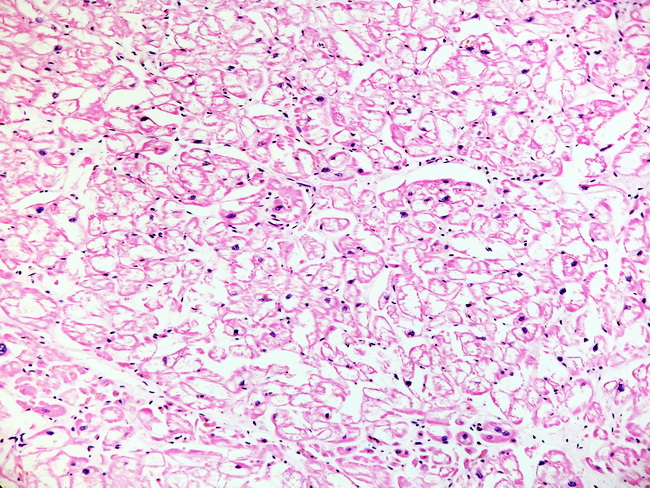
Genetics: Fabry disease is caused by mutations in α-galactosidase A gene on Xq22. 1. The mutation prevalence is estimated to be between 1:40,000 to 1:117,000 live male births. More than 500 mutations have been identified; most are family specific. About 5% of cases are sporadic. Clinical Features: Hemizygous males with little or no α-galactosidase A activity have the most severe multisystem manifestations and present early in childhood or adolescence. Clinical features include painful paresthesias in lower extremities, diffuse angiokeratomas, hypohidrosis or anhidrosis, corneal opacities and cataracts. Renal manifestations begin in the 2nd decade of life with proteinuria, hematuria, and hypertension and progress to renal failure by 3rd or 4th decade of life. Males with residual α-galacrosidase A activity present in 4th or 5th decade of life with cardiac manfestations (chest pain, arrhythmia, sudden death) or renal disease (proteinuria, hypertension, renal failure). Heterozygous females are carriers and are asymptomatic or develop mild symptoms, especially renal insufficiency, later in life. Diagnosis can be made by measuring α-galactosidase A in leukocytes and urine and by next-generation sequencing to detect specific mutations. About this image: Low magnification view of heart section showing hypertrophy and prominent cytoplasmic vacuolization of cardiac myocytes. Image courtesy of: Dr. Esther Youd, Autopsy Pathologist, Univ. of Glasgow, Glasgow, Scotland; used with permission.