Apr 2020
Glucagonoma
Reviewer(s): Dharam Ramnani, MD
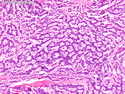
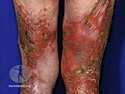
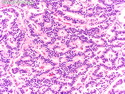
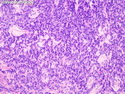
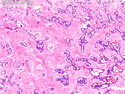
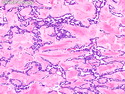
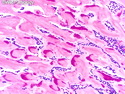
GLUCAGONOMA: It is the 4th most common functioning pancreatic neuroendocrine tumor (after insulinoma, gastrinoma, and VIPoma) and accounts for 1-2% of all PanNETs. Uncontrolled glucagon secretion by the tumor causes a distinctive syndrome of glucose intolerance, anemia, weight loss, depression, venous thromboembolism, and skin rash (necrolytic migratory erythema). Fasting plasma glucagon levels are between 550 and 6600 pg/ml (normal < 200 pg/ml). It is usually sporadic; about 13%-17% are associated with MEN type 1. Most cases are malignant. Liver metastases are seen in 50-70% of patients at initial presentation. About 75% of cases are ultimately fatal. Grossly, glucagonomas are solitary large tumors measuring 5 to 10 cms in size. Microscopic features are similar to those seen in other PanNETs. Mitotic activity and cytologic atypia are not prominent. Immunohistochemically, glucagon and other markers like chromogranin, synaptophysin, and PP are positive. Treatment consists of symptomatic relief, restoring nutritional status and controlling hyperglycemia to stabilize the patient before surgery. Treatment of choice for non-metastatic tumors is surgical resection. Unresectable tumors are treated with somatostatin analogs such as octreotide and lanreotide or mTOR inhibitors such as everolimus. The 5-yr survival rate after surgical resection is 70% for non-metastatic tumors. The overall cure rate is only about 25%. GLUCAGON CELL HYPERPLASIA & NEOPLASIA: A rare autosomal recessive benign condition with elevated glucagon levels without the glucagonoma syndrome. Germline mutations in GCGR gene on chromosome 17q25.3 are seen in 50% of cases.