Mar 2017
Neurofibromatosis 1
Reviewer(s): Dharam M. Ramnani, MD
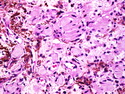
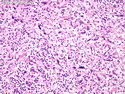
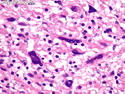
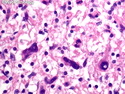
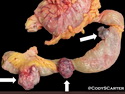
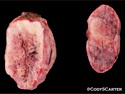
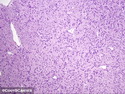
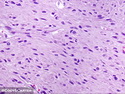
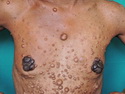
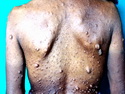
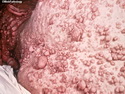
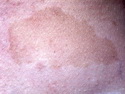
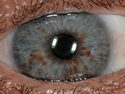
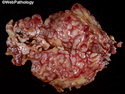
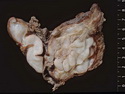
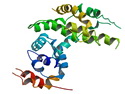
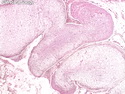
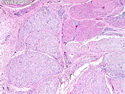
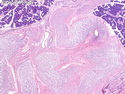
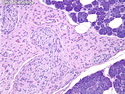
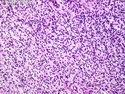
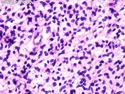
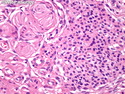
Neurofibromatosis 1 (NF1) belongs to a group of inherited cancer predisposition syndromes caused by germline mutations in genes of RAS/Mitogen-Activated Protein Kinase (MAPK) pathway. They are collectively known as RASopathies. NF1 (previously known as von Recklinghausen disease) is autosomal dominant with very high penetrance and affects about 1 in 3000 individuals. NF1 is caused by genetic defects in the tumor suppressor gene NF1 on chromosome 17q11.2. NF1 codes for Neurofibromin that is expressed in brain and peripheral nerves. It is a RAS GTPase-activating protein that inactivates RAS proteins. Loss of NF1 function promotes RAS activity which activates downstream signaling pathways leading to tumor formation. NF1 is characterized by multiple cafe-au-lait spots and neurofibromas (usually plexiform or diffuse type). Other manifestations include: CNS lesions (optic nerve glioma, astrocytoma, heterotopias), pigmented hamartoma of iris (Lisch nodule), skeletal abnormalities, vascular abnormalities (renovascular hypertension), gynecomastia, non-neural tumors (pheochromocytoma, myelogenous leukemia), and GIST. Variant forms include: Segmental neurofibromatosis (neurofibromas localized to one area (segment) of body; Gastrointestinal NF; Familial Spinal NF; and Familial cafe-au-lait spots. Transformation to malignant peripheral nerve sheath tumor (MPNST) is only seen in plexiform neurofibromas. The lifetime risk of MPNST in NF1 patients is 8% to 13%. Patients with multiple deep-seated plexiform neurofibromas and those with large deletions in NF1 gene are at a greater risk. References: 1) Enzinger & Weiss's Soft Tissue Tumors, Sixth Edition, 2014; p. 800-813. Updated: April 2, 2017
.jpg)