
Hirschsprung Disease



Comments:
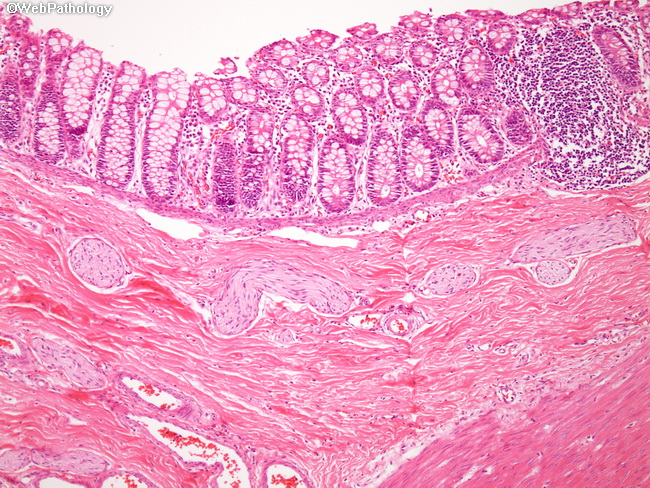
Hirschsprung disease is characterized by the absence of parasympathetic ganglion cells in both submucosal (Meissner) as well as myenteric (Auerbach) plexuses in the distal gastrointestinal tract. The most likely cause is failure of vagal neural crest cells to migrate into the involved segments of the bowel between 5 and 12 weeks of gestation. The denervated colon lacks coordinated peristaltic activity and undergoes persistent contraction causing functional obstruction. The normally innervated proximal colon becomes massively dilated (megacolon). It typically presents in term neonates with failure to pass meconium in the 1-2 days after birth. In addition, there is often abdominal distension, vomiting, and visible, ineffective peristalsis. Fluid and electrolyte imbalance can develop quickly. Massive dilation and thinning of the colonic wall can cause perforation, usually near the cecum, followed by peritonitis. The incidence of Hirschsprung disease is 1 in 5000 live births with male predominance. About 10% of cases occur in children with Down syndrome. Additional 5% of patients have other serious neurologic defects. This image is from a suction biopsy of the rectum showing absence of ganglion cells in the submucosal plexus as well as the presence of hypertrophic submucosal nerve fibers. The affected segments may also show mucosal inflammation and shallow ulcers (both features are absent here).