Apr 2020
Pancreatoblastoma
Reviewer(s): Dharam Ramnani, MD
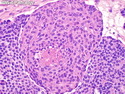
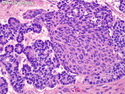
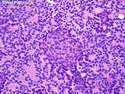
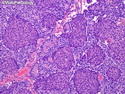
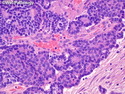
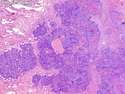
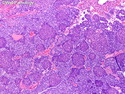
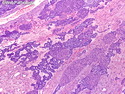
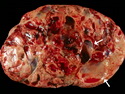
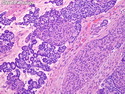
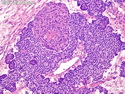
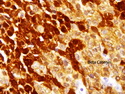
Pancreatoblastoma is a rare malignant pancreatic tumor that is usually seen in children (mean age at diagnosis 4-5 years). Most cases are sporadic. Some are associated with Beckwith-Wiedemann syndrome (BWS) or familial adenomatous polyposis (FAP). It may be discovered incidentally or present with abdominal pain, abdominal mass, weight loss, diarrhea, and nausea. Serum AFP, CEA, and ACTH are frequently elevated. More than 80% of cases show loss of 11p. Mutations in CTNNB1 occur in 50-80% of cases resulting in nuclear and cytoplasmic accumulation of β-catenin. Cases associated with FAP show mutations in the APC gene. Abnormalities in TP53 and KRAS are not seen. They are large (mean size 10 cm), solid, and well-circumscribed tumors that may be partially encapsulated. Cases associated with BWS are mostly cystic. The tumor shows acinar, ductal, endocrine, and squamoid differentiation. There is no cytologic atypia. Mitotic activity is variably increased. The tumor cells are positive for acinar markers (trypsin, chymotrypsin, lipase), BCL10, neuroendocrine markers (synaptophysin, chromogranin A), and ductal markers (CK7, CK19). α-fetoprotein, α1-antitrypsin, and CEA are also positive. Pancreatoblastomas are indolent tumors but they can be locally invasive and metastasize. About 35% of patients have metastases (liver, regional lymph nodes) at presentation. The prognosis is excellent if the tumor is resected before it has metastasized. The overall cure rate is about 50%.
.jpg)