
Angiosarcoma : Introduction


Comments:
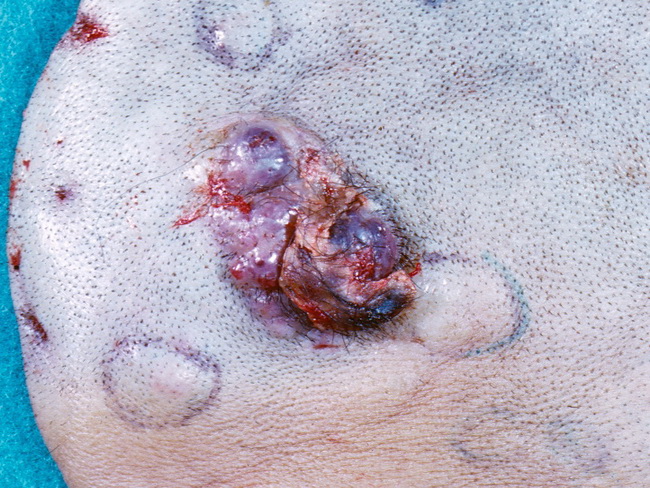
Introduction: Angiosarcomas are rare mesenchymal tumors showing endothelial differentiation and comprise less than 1% of all sarcomas. They show variable degrees of vascular formation and range from well-differentiated tumors resembling hemangioma to highly anaplastic solid tumors that may be mistaken for poorly-differentiated carcinomas or sarcomas. They are no longer separated into hemangiosarcomas and lymphangiosarcomas. Angiosarcomas are highly aggressive tumors with a poor prognosis. Locations: Tumors arising in skin and superficial soft tissues (cutaneous angiosarcomas) make up about 50% of cases. Another 40% of cases involve parenchymal organs (breast, heart, spleen, liver, bone). The remaining 10% occur in deep soft tissues. The peak incidence is seen in the 7th decade. Angiosarcomas are rare in children; but when they do occur, they tend to involve deep soft tissues of head and neck, mediastinum, and internal organs more frequently than in adults. Clinical Subtypes: On the basis of clinical presentation, pathogenesis, and prognosis, angiosarcomas are subdivided into several groups:
- Cutaneous Angiosarcoma (unassociated with radiation or lymphedema) - scalp, face, forehead, and neck
- Lymphedema-associated Angiosarcoma (including Stewart-Treves Syndrome)
- Post-radiation Angiosarcoma
- Angiosarcoma of Parenchymal Organs (breast, heart, liver, spleen, bones)
- Angiosarcoma of Deep Soft Tissues